Molecular dynamics (MD) is a popular method for studying molecular systems and microscopic processes at the atomic level. However, MD simulations can be quite computationally expensive due to the intricate temporal and spatial resolutions needed. Due to the computing load, much research has been done on alternate techniques that can speed up simulation without sacrificing accuracy. Creating surrogate models based on deep learning is one such strategy that can effectively replace conventional MD simulations.
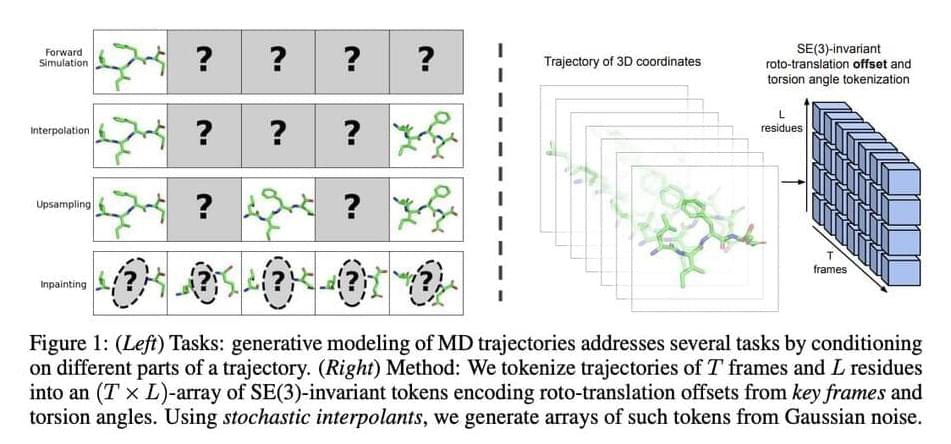
In recent research, a team of MIT researchers introduced the use of generative modeling to simulate molecular motions. This framework eliminates the need to compute the molecular forces at each step by using machine learning models that are trained on data obtained by MD simulations to provide believable molecular paths. These generative models can function as adaptable multi-task surrogate models, able to carry out multiple crucial tasks for which MD simulations are generally employed.
These generative models can be trained for a variety of tasks by carefully choosing and conditioning on specific frames of a molecule trajectory. These tasks include the following.